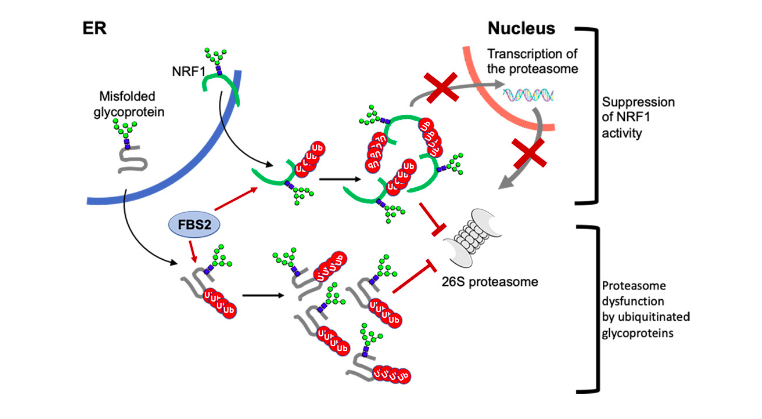
Image: A schematic model of the mechanism of proteasome dysfunction by FBS2 in NGLY1 deficiency. FBS2 induces cellular toxicity in the absence of NGLY by two steps. First, FBS2 ubiquitinates misfolded glycoprotein substrates. Ubiquitinated glycoproteins recruited to the proteasome can strongly inhibit the proteasome activity, because of the presence of N-glycans. Accumulation of ubiquitinated glycoproteins results in the inhibition of the proteasome. Cells sense the inhibition of the proteasome, they try to induce transcriptional activation of proteasome subunits by active NRF1. But FBS2 suppresses by inhibiting nuclear import of NRF1. Moreover, induced NRF1 also causes proteasome dysfunction as the ubiquitinated glycoprotein substrates.
Peptide: N-glycanase (NGLY1) is an evolutionarily conserved enzyme for removing N-linked glycans (N-glycans) from glycoproteins and is involved in proteostasis of N-glycoproteins in the cytosol. In 2012, a rare genetic disorder called NGLY1 deficiency was discovered by an exome analysis. Symptoms in patients with NGLY1 deficiency include global developmental delay, hypotonia, hypo/alacrima, movement disorder, scoliosis, abnormal liver, brain functions and peripheral neuropathy. Unfortunately, no therapeutic treatment is currently available to this devastating disease, because the exact mechanism of harm is not yet understood. So far, Dr Suzuki’s team in RIKEN and T-CiRA have developed various animal models for NGLY1 deficiency and reported that NGLY1-knockout in B6 mice is embryonically lethal.
In the cytosol, there are other proteins involved in processing of N-glycoproteins. One such group of proteins is glycoprotein-specific ubiquitin ligase subunits, FBS (F-box proteins recognizing sugar chains), which ubiquitinates unfolded glycoproteins in the cytosol. Ubiquitination of proteins serves as signals for proteolysis by the proteasome, a proteolytic machinery essential for cellular proteostasis and health. The researchers found that deletion of gene encoding FBS2, one of FBS proteins, restored the lethality of NGLY1-KO B6 mice and the FBS2;NGLY1 double-KO mice grew without apparent abnormalities. This finding reveals that the activity of FBS2 could cause the lethality in the embryos of NGLY1-KO mice.
The detrimental effects of FBS2 in the absence of NGLY1 were also observed in culture cells. The researchers noticed that FBS2 overexpression in NGLY1-KO cell lines suppressed cell growth and eventually induced cell death. FBS2 ubiquitinated glycoproteins that failed to remove N-glycans in the absence of NGLY1, and the ubiquitinated glycoproteins were not degraded and accumulated in cells. Ubiquitinated proteins are normally degraded by the proteasome, but ubiquitinated glycoproteins induced proteasome inhibition. The prolonged proteasome inhibition leads to cell death. In order to maintain proteasome function, cells induce the synthesis of new proteasomes by the transcriptional factor NRF1 upon proteasome dysfunction. Interestingly, NRF1 is a glycoprotein that resides in the endoplasmic reticulum and degraded by the proteasome under normal conditions. When proteasome activity is suppressed, NRF1 is stabilized and translocated into the nucleus where it activates transcription of the proteasome. However, in the absence of NGLY1, FBS2 ubiquitinated NRF1 and suppressed its transcriptional activity of the proteasome. Thus, FBS2 inhibited proteasome activity and also injured the recovery function of the proteasome in loss of NGLY1. The presence of ubiquitinated NRF1 was also observed in NGLY1-KO mouse embryos.
This work paves the way to development of therapeutics for this intractable disease.
Source:
Yoshida, Y., et al. (2021) Loss of peptide:N-glycanase causes proteasome dysfunction mediated by a sugar-recognizing ubiquitin ligase. Proceedings of National Academy of Sciences. doi.org/10.1073/pnas.2102902118