A new genetic cause of Congenital Disorders of Glycosylation (CDG) has been identified, according to a recent study in The American Journal of Human Genetics. This means a blood sample can now confirm diagnosis of this form of CDG, notes Andrew C. Edmondson, MD, PhD, an attending physician in Human Genetics and Metabolism at Children’s Hospital of Philadelphia and co-author on the study. Edmondson is one of the founding members of Frontiers in Congenital Disorders of Glycosylation (FCDGC).
People born with CDG can experience a range of neurological symptoms, developmental issues, growth struggles, and organ problems. Due to the fact that CDG represent a variety of rare disorders with such a range of presentations, children with CDG are often misdiagnosed.
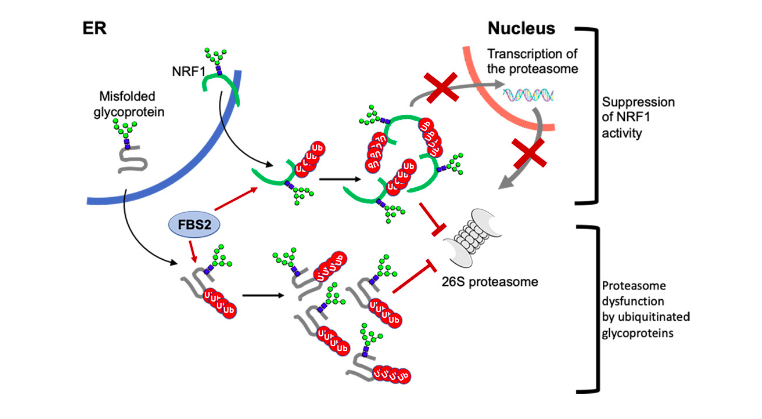
Using a combination of exome sequencing and gene matching, researchers identified 12 individuals from seven families who present with CDG with a variant in EDEM3, a gene which encodes a protein responsible for recognizing misfolded glycoproteins in the endoplasmic reticulum (ER). The presence of misfolded ER proteins is involved in several CDGs, so tracing this system was important.
Individuals in the study affected by CDG showed either homozygous or compound heterozygous variants in the EDEM3 gene. Unaffected parents were all heterozygous carriers. People from two families carried a common, identical region of homozygosity (with no records of inter-familial consanguinity); both families were of Portuguese Romani origin. A control group of 96 healthy control subjects in the Portuguese Romani population were also screened for this variant, with one heterogeneous allele detected. It’s possible links to this population may represent a founder effect.
The CDG patients in the study presented with developmental delay with intellectual delay and speech delay in some cases. Half showed hypotonia and many presented with dysmorphic facial features.
By comparing ratios of glycosylation abnormalities across affected CDG patients, researchers found a global impact on N-glycosylation in EDEM3 deficiency and suggest among EDEM3-CDG-affected people, abnormal N-glycan profiling patterns provide additional diagnostic biomarkers.
As Edmondson notes, EDEM3 knock out mice “had similar biochemical abnormalities in blood and brain as were seen in the human sample.” Now that the gene has been identified, future research is likely to focus upon how loss of EDEM3 activity leads to disease and this set of symptoms that are so often misunderstood or misdiagnosed.
Source:
Rare Diseases Clinical Research Network
Journal Reference:
Polla, D.L., et al. (2021) Bi-allelic variants in the ER quality-control mannosidase gene EDEM3 cause a congenital disorder of glycosylation. American Journal of Human Genetics. 10.1016/j.ajhg.2021.05.010